Last week the supervisor of my lab actually decided for me to work on a different project that still connects with the previous project I was working on. Previously I was supposed to express and eventually purify the telomerase RNA binding domain(TRBD). In order to purify any protein, the protein has to have a specific tag that would help select for that only that protein. In this case, the TRBD protein was tagged with a maltose binding protein. However after purification, the maltose binding protein must be separated from the TRBD protein. In order to do this a specific protease is used. In this case a TEV protease encoded by a tobacco etch virus cleaves at specific site to separate the maltose binding protein from TRBD. This digestion process usually takes about a day. This image I got from https://www.neb.com/applications/protein-expression-and-purification/coupled-protein-expression-and-purification/maltose-binding-protein-expression may better describe what I am talking about.
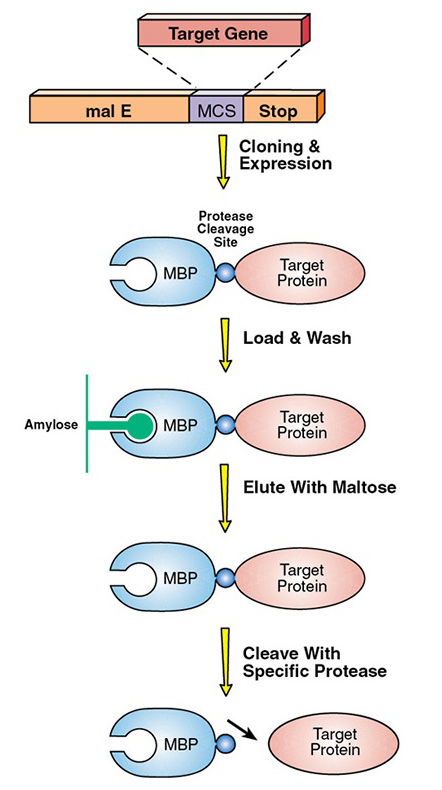
The TEV protease binds to and cleaves off a specific amino acid sequence located between TRBD and the maltose binding protein. The supervisor in my lab recently read about a single amino acid change in this sequence may allow the TEV protease to cleave off the sequence faster. This would be more efficient for purifying the TRBD protein because the digestion process may happen in less than two hours vs. an entire day. My project right now is to show how efficient the single amino acid change in the TEV cleavage sequence would be for purifying TRBD because the lab has never tested it before.
Because of this sudden project change the only hands on thing I actually did was make a buffer for dispensing the pellets containing TRBD as part of the purification for TRBD. The rest of the time I attended lab meetings where each person talked about their progress with their projects and when the supervisor tried to explain this project to me.
This week I will be starting the new project and in the next post I will talk more about it in detail. Like some other people, I may also have to change the title of my presentation.
The TEV protease binds to and cleaves off a specific amino acid sequence located between TRBD and the maltose binding protein. The supervisor in my lab recently read about a single amino acid change in this sequence may allow the TEV protease to cleave off the sequence faster. This would be more efficient for purifying the TRBD protein because the digestion process may happen in less than two hours vs. an entire day. My project right now is to show how efficient the single amino acid change in the TEV cleavage sequence would be for purifying TRBD because the lab has never tested it before.
Because of this sudden project change the only hands on thing I actually did was make a buffer for dispensing the pellets containing TRBD as part of the purification for TRBD. The rest of the time I attended lab meetings where each person talked about their progress with their projects and when the supervisor tried to explain this project to me.
This week I will be starting the new project and in the next post I will talk more about it in detail. Like some other people, I may also have to change the title of my presentation.

